If you've done an autosomal DNA test for genealogy (e.g. AncestryDNA, 23andMe, FamilyTreeDNA's Family Finder, or MyHeritage), you may have gotten a bewildering collection of DNA matches that the testing company suggests are some sort of cousin. For example, when I downloaded my full set of AncestryDNA results last week, I had 14,435 matches. That's a lot of relatives!
In January 2017, Blaine Bettinger (who blogs as The Genetic Genealogist) published "The Danger of Distant Matches". In this post, he compared his matches to those of both of his parents and found that 35% of his matches were not shared with either of his parents. The vast majority of these problematic matches shared a small amount of DNA (10 centimorgans (cM)* or less). Since he got all of his DNA from his parents, this means that these matches have to be false positives (or false negatives for his parents).
Last month, I had the privilege of studying with Bettinger and other experts in the week-long course on Practical Genetic Genealogy at the Genealogical Research Institute of Pittsburgh (GRIP). We discussed the distant matches findings and Bettinger commented that he'd like to have more people do this analysis so we can have a bigger data set to consider.** Since both of my parents have also tested at AncestryDNA (and given their permission for me to blog about their results), I replicated Bettinger's process with my own family's data.
Using the DNAGedcom Client, I downloaded match lists for me and both of my parents on July 26 and July 27, 2017. I used the Match-o-matic tool, available in Version 1.5.1.3 of the DNAGedcom Client, to analyze the match lists to see which matches are (and are not) shared.
Overall, 17% of my matches were unshared with either parent (2,411 out of 14,435), which is more encouraging than the 32% rate that Bettinger found for his results.
Here's the breakdown for the categories that Bettinger used:
Largest match: 18.1 cM
10 share 15 cM or more
130 share 10 cM or more (5% of unshared matches)
894 share 7-10 cM (37% of unshared matches)
1387 share 6-7 cM (58% of unshared matches)
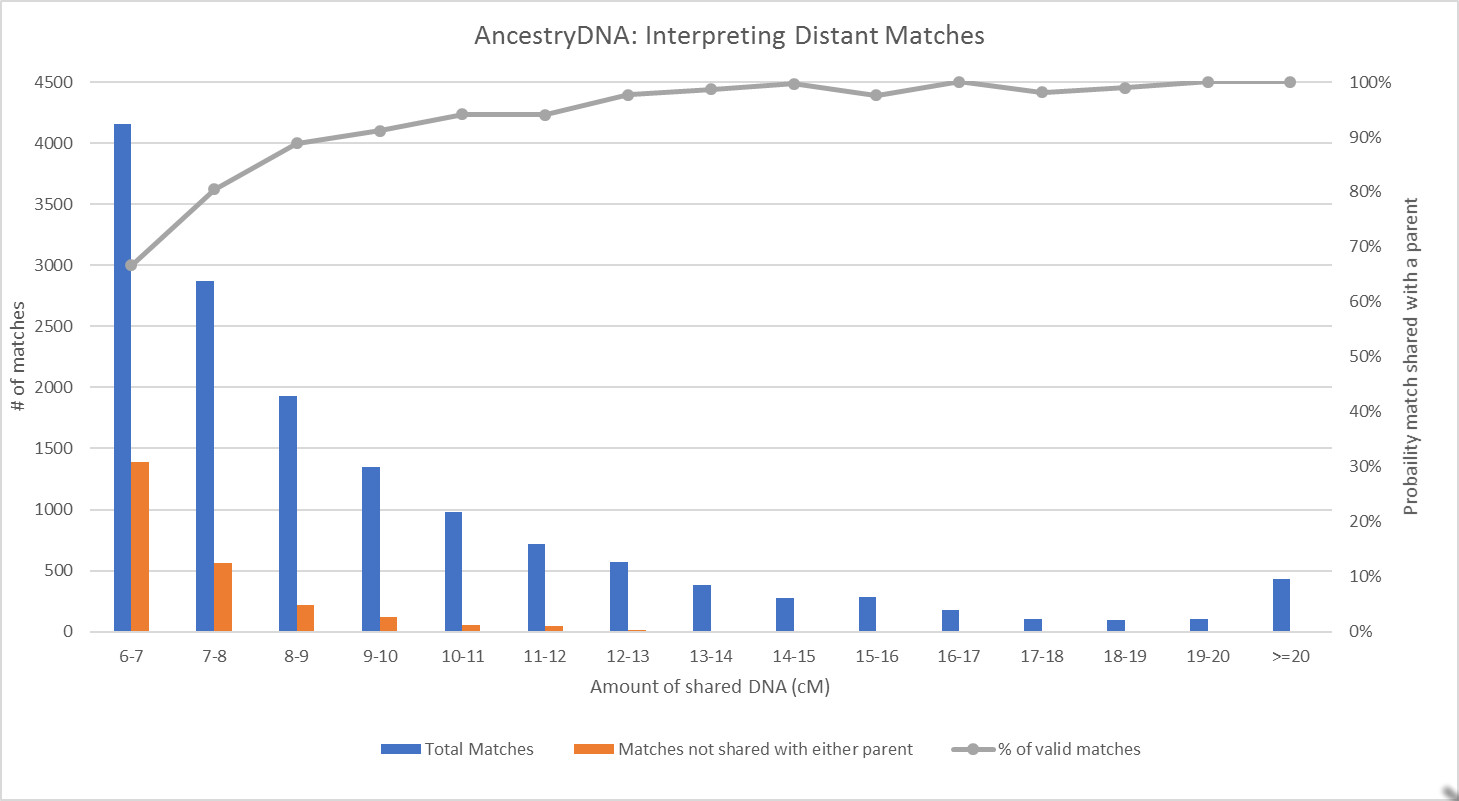
I tend to like visual representations of data, so I wanted to see the shape of the curve for the proportion of matches that appear to be valid. I put the data into buckets based on the total amount of shared DNA in centimorgans and here's what I found:

From this set of data, it appears that the probability of one of my matches being shared with a parent is very good (98% or better) for matches down to about 12 cM. When matches share less DNA with me than that, the likelihood that they also show up as a match for one of my parents starts to drop off. By the time we get down to about 9 cM, the probability of a match being shared with a parent drops to about 90%. Below that, it drops off more dramatically, with only two-thirds (67%) of matches between 6-7 cM being shared with one of my parents.
So what does this mean? Should you ignore all matches below a certain threshold because they might be, in Bettinger's words, poison? Is there some threshold above which you can say with total certainty that the match is real?
As with most things, I think a set of context-sensitive guidelines rather than hard-and-fast rules are the way to go. Here are some of the guidelines that I use when analyzing autosomal data:
- Start with closer matches, rather than more distant ones. Matches that share more DNA are not only more likely to be valid matches, but they're also likely to be more closely related, so it may be easier to figure out who the shared ancestors are.
- Use caution when considering distant matches. Perhaps because my results were a bit more encouraging than Bettinger's (or perhaps because I haven't been at this as long as he has which may lead to some naiveté on my part), I might be more favorably inclined to consider matches below the 15 cM guideline that Bettinger describes as the "safe zone". However, it is clear that the smaller the amount of DNA the matches share, the higher the risk that the match might not be valid.
- Correlate multiple sources of evidence! The amount of DNA shared with a match is only one piece of the puzzle. The best way I know of to determine whether a match is valid or not is to correlate the amount shared DNA with other available evidence. How long are the matching segment(s)? Do you and this match share other matches in common? If so, who are they and what do you know about them? Does this match have a tree available? If so, how complete is it? How complete is your own tree? This is just a subset of the questions I would ask when analyzing any match.
In practice, I tend to incorporate small matches into my analysis when they come to my attention for some other reason. A distant match may fit into a triangulation group (a group of three or more people, not closely related to one another, who share a particular DNA segment in common), may have an enticing set of in-common-with matches related to a question I'm researching, or may capture my attention due to a tree with surname(s) or location(s) that give me a clue as to where this match might fit in with the questions I'm trying to answer.
For those of you who are digging into autosomal DNA, what guidelines do you use to decide when to pay attention to a distant match?
*A centimorgan (cM) is a measure of the probability of a recombination event happening between two locations on DNA. Because recombination is more likely in some locations on the genome than others, it doesn't correlate perfectly with number of base pairs, or rungs in the ladder of DNA. However, for the purposes of this discussion you can think of it more-or-less as a measure of distance. On average, a centimorgan corresponds to about 1 million base pairs.
**I know of at least one other person who has also published a similar analysis: http://www.irelanddavis.com/dna/articles/parents/Default.aspx. I was glad to have two sets of data other than my own to look at!